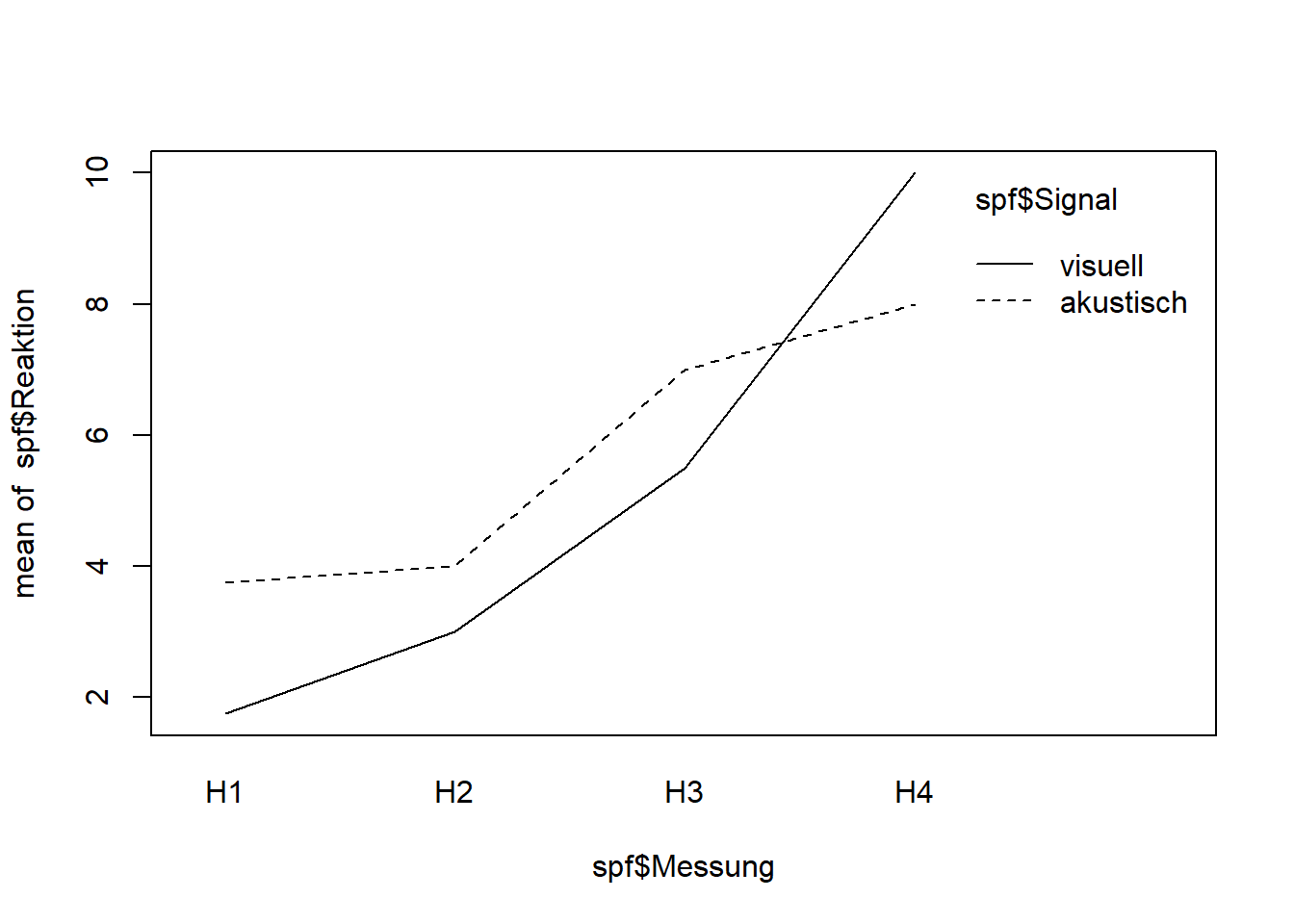
# LM mit random intercept = einfaches LMMspf.aov <-aov(Reaktion ~ Signal * Messung +Error(VP), data = spf)summary(spf.aov)
Error: VP
Df Sum Sq Mean Sq F value Pr(>F)
Signal 1 3.125 3.125 2 0.207
Residuals 6 9.375 1.562
Error: Within
Df Sum Sq Mean Sq F value Pr(>F)
Messung 3 194.50 64.83 127.89 2.52e-12 ***
Signal:Messung 3 19.37 6.46 12.74 0.000105 ***
Residuals 18 9.13 0.51
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
# Interaktion anschaueninteraction.plot(spf$Messung, spf$Signal, spf$Reaktion)# Nun als LMMlibrary("nlme")
# Mit random intercept (VP) und random slope (Messung)spf.lme.1<-lme(Reaktion ~ Signal * Messung, random =~ Messung | VP, data = spf)# Nur random interceptspf.lme.2<-lme(Reaktion ~ Signal * Messung, random =~1| VP, data = spf)# Modelle anschauenanova(spf.lme.1)
-> Hirschparasitenbeispiel aus Kapitel 13 von Zuur u. a. (2009)
# Daten laden und für GLMM aufbereitenDeerEcervi <-read_delim("datasets/stat5-8/DeerEcervi.txt", "\t", col_types =cols("Farm"="f"))# Daten anschauenhead(DeerEcervi)
# A tibble: 6 × 4
Farm Sex Length Ecervi
<fct> <dbl> <dbl> <dbl>
1 AL 2 164 0
2 AL 1 216 0
3 AL 1 208 0
4 AL 1 206 14.4
5 AL 1 204 0
6 AL 1 200 0
summary(DeerEcervi)
Farm Sex Length Ecervi
MO :209 Min. :1.000 Min. : 75.0 Min. : 0.00
CB : 85 1st Qu.:1.000 1st Qu.:151.0 1st Qu.: 0.00
QM : 60 Median :1.000 Median :163.0 Median : 6.60
BA : 50 Mean :1.458 Mean :161.8 Mean : 45.42
PN : 37 3rd Qu.:2.000 3rd Qu.:174.9 3rd Qu.: 35.79
MB : 34 Max. :2.000 Max. :216.0 Max. :2186.60
(Other):351
# Anzahl Larven hier in Presence/Absence übersetztDeerEcervi$Ecervi.01<- DeerEcervi$EcerviDeerEcervi$Ecervi.01[DeerEcervi$Ecervi >0] <-1# Numerische Geschlechtscodierung als FactorDeerEcervi$fSex <-as.factor(DeerEcervi$Sex)# Hischlänge standardisierenDeerEcervi$CLength <- DeerEcervi$Length -mean(DeerEcervi$Length)
-> Nun sind die Daten bereit:
Die Parasitenbefalldaten wurden in Parasiten Präsenz/Absenz (1/0) übersetzt.
Die Hirschlänge wurde standardisiert, damit der Achsenabschnitt (intercept) des Modells interpretierbar ist, standardisierte entspricht nun der Achsenabschnitt einem durschnittlich langen Hirsch.
# Zunächst als GLM# Interaktionen mit fFarm nicht berücksichtigt, da zu viele Freiheitsgrade verbraucht würdenDE.glm <-glm(Ecervi.01~ CLength * fSex + Farm, family = binomial, data = DeerEcervi)drop1(DE.glm, test ="Chi")
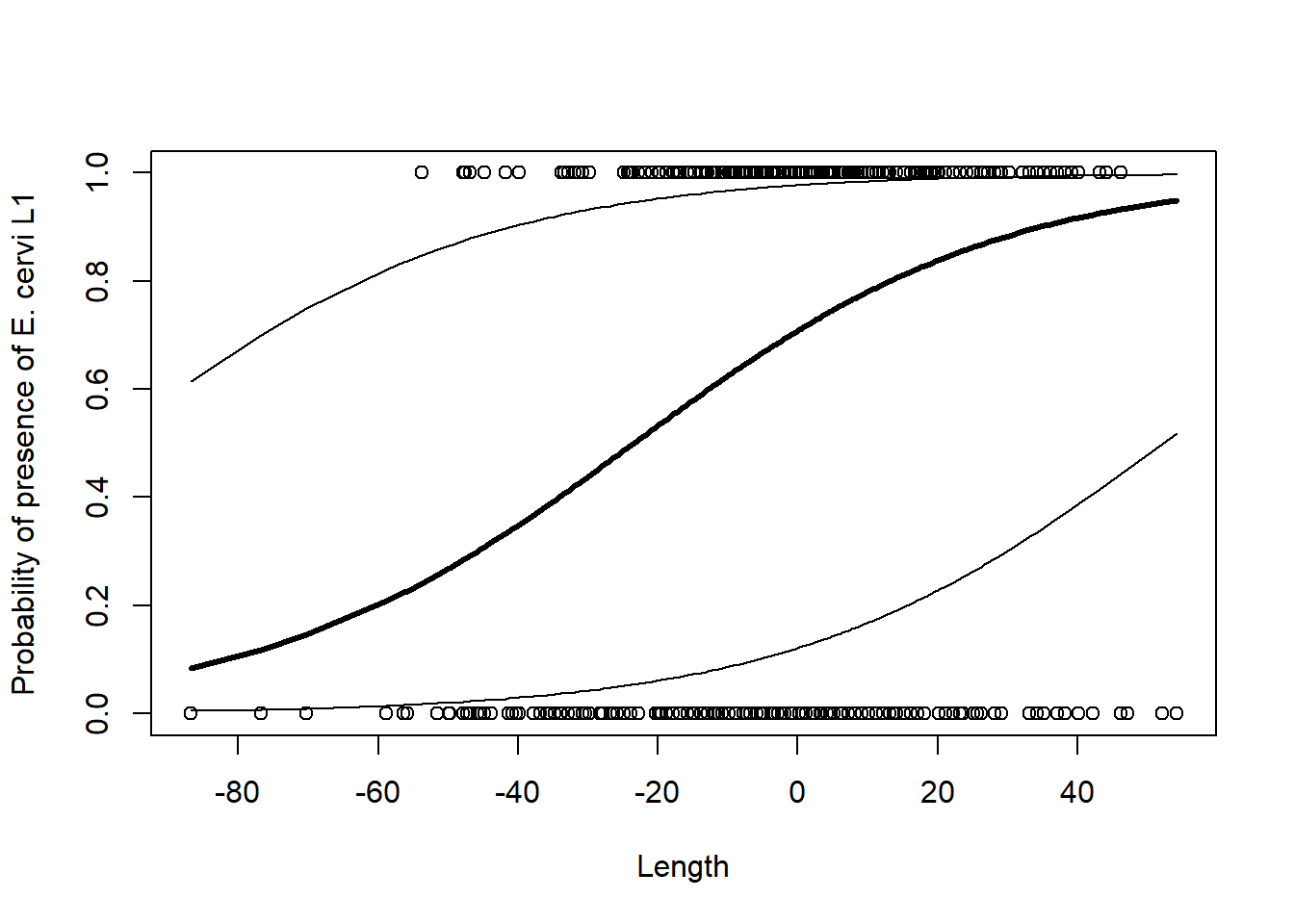
# Modellvoraussagen berechnen# So könnte direkt auf die coefficients zugegriffen werden: DE.PQL$coefficients[[1]][1]g <-0.8883697+0.0378608* DeerEcervi$CLength# Rücktransformieren aus Logitp.averageFarm1 <-exp(g) / (1+exp(g))# Sortierung der Hirschgrössen für's PlottenI <-order(DeerEcervi$CLength)# Plottenplot(DeerEcervi$CLength, DeerEcervi$Ecervi.01,xlab ="Length",ylab ="Probability of presence of E. cervi L1")lines(DeerEcervi$CLength[I], p.averageFarm1[I], lwd =3)# Vertrauensintervalle (CI) mit SD von Random factor berechnen# Generell CI = mean + 1.96*SDp.Upp <-exp(g +1.96*1.462108) / (1+exp(g +1.96*1.462108))p.Low <-exp(g -1.96*1.462108) / (1+exp(g -1.96*1.462108))lines(DeerEcervi$CLength[I], p.Upp[I])lines(DeerEcervi$CLength[I], p.Low[I])
# Dasselbe mit dem lme4-Packagelibrary("lme4")DE.lme4 <-glmer(Ecervi.01~ CLength * fSex + (1| Farm),family = binomial, data = DeerEcervi)summary(DE.lme4)
Generalized linear mixed model fit by maximum likelihood (Laplace
Approximation) [glmerMod]
Family: binomial ( logit )
Formula: Ecervi.01 ~ CLength * fSex + (1 | Farm)
Data: DeerEcervi
AIC BIC logLik deviance df.resid
832.6 856.1 -411.3 822.6 821
Scaled residuals:
Min 1Q Median 3Q Max
-6.2678 -0.6090 0.2809 0.5022 3.4546
Random effects:
Groups Name Variance Std.Dev.
Farm (Intercept) 2.391 1.546
Number of obs: 826, groups: Farm, 24
Fixed effects:
Estimate Std. Error z value Pr(>|z|)
(Intercept) 0.938969 0.356004 2.638 0.00835 **
CLength 0.038964 0.006917 5.633 1.77e-08 ***
fSex2 0.624487 0.222938 2.801 0.00509 **
CLength:fSex2 0.035859 0.011409 3.143 0.00167 **
---
Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
Correlation of Fixed Effects:
(Intr) CLngth fSex2
CLength -0.107
fSex2 -0.189 0.238
CLngth:fSx2 0.091 -0.514 0.232
library("glmmML")DE.glmmML <-glmmML(Ecervi.01~ CLength * fSex,cluster = Farm, family = binomial, data = DeerEcervi)summary(DE.glmmML)
Call: glmmML(formula = Ecervi.01 ~ CLength * fSex, family = binomial, data = DeerEcervi, cluster = Farm)
coef se(coef) z Pr(>|z|)
(Intercept) 0.93968 0.357915 2.625 8.65e-03
CLength 0.03898 0.006956 5.604 2.10e-08
fSex2 0.62451 0.224251 2.785 5.35e-03
CLength:fSex2 0.03586 0.011437 3.135 1.72e-03
Scale parameter in mixing distribution: 1.547 gaussian
Std. Error: 0.2975
LR p-value for H_0: sigma = 0: 1.346e-41
Residual deviance: 822.6 on 821 degrees of freedom AIC: 832.6
Zuur, A., E. N. Ieno, N. Walker, A. A. Saveliev, und G. M. Smith. 2009. Mixed Effects Models and Extensions in Ecology with R. Statistics for Biology and Health. Springer New York. https://books.google.ch/books?id=vQUNprFZKHsC.
Quellcode
---date: 2023-11-13lesson: Stat5thema: Von linearen Modellen zu GLMMsindex: 1format: html: code-tools: source: trueknitr: opts_chunk: collapse: false---# Stat5: Demo- Download dieses Demoscript via "\</\>Code" (oben rechts)- Datensatz *spf.csv*- Datensatz *DeerEcervi.txt*## Split-plot ANOVAReaktionszeitenbeispiel aus Kapitel 14 von @logan2010```{r}# Daten ladenlibrary("readr")spf <-read_delim("datasets/stat5-8/spf.csv", ";")# Daten anschauenhead(spf)# LM mit random intercept = einfaches LMMspf.aov <-aov(Reaktion ~ Signal * Messung +Error(VP), data = spf)summary(spf.aov)# Interaktion anschaueninteraction.plot(spf$Messung, spf$Signal, spf$Reaktion)# Nun als LMMlibrary("nlme")# Mit random intercept (VP) und random slope (Messung)spf.lme.1<-lme(Reaktion ~ Signal * Messung, random =~ Messung | VP, data = spf)# Nur random interceptspf.lme.2<-lme(Reaktion ~ Signal * Messung, random =~1| VP, data = spf)# Modelle anschauenanova(spf.lme.1)anova(spf.lme.2)summary(spf.lme.1)summary(spf.lme.2)```## GLMM-\> Hirschparasitenbeispiel aus Kapitel 13 von @zuur2009```{r}# Daten laden und für GLMM aufbereitenDeerEcervi <-read_delim("datasets/stat5-8/DeerEcervi.txt", "\t", col_types =cols("Farm"="f"))# Daten anschauenhead(DeerEcervi)summary(DeerEcervi)# Anzahl Larven hier in Presence/Absence übersetztDeerEcervi$Ecervi.01<- DeerEcervi$EcerviDeerEcervi$Ecervi.01[DeerEcervi$Ecervi >0] <-1# Numerische Geschlechtscodierung als FactorDeerEcervi$fSex <-as.factor(DeerEcervi$Sex)# Hischlänge standardisierenDeerEcervi$CLength <- DeerEcervi$Length -mean(DeerEcervi$Length)```-\> Nun sind die Daten bereit:- Die Parasitenbefalldaten wurden in Parasiten Präsenz/Absenz (1/0) übersetzt.- Die Hirschlänge wurde standardisiert, damit der Achsenabschnitt (intercept) des Modells interpretierbar ist, standardisierte entspricht nun der Achsenabschnitt einem durschnittlich langen Hirsch.```{r}# Zunächst als GLM# Interaktionen mit fFarm nicht berücksichtigt, da zu viele Freiheitsgrade verbraucht würdenDE.glm <-glm(Ecervi.01~ CLength * fSex + Farm, family = binomial, data = DeerEcervi)drop1(DE.glm, test ="Chi")summary(DE.glm)anova(DE.glm)# Response curves für die einzelnen Farmen (Weibliche Tiere: fSex = "1" )plot(DeerEcervi$CLength, DeerEcervi$Ecervi.01,xlab ="Length", ylab ="Probability of \ presence of E. cervi L1")I <-order(DeerEcervi$CLength)AllFarms <-unique(DeerEcervi$Farm)for (j in AllFarms) { mydata <-data.frame(CLength = DeerEcervi$CLength, fSex ="1",Farm = j ) n <-dim(mydata)[1]if (n >10) { P.DE2 <-predict(DE.glm, mydata, type ="response")lines(mydata$CLength[I], P.DE2[I]) }}# glmmlibrary("MASS")DE.PQL <-glmmPQL(Ecervi.01~ CLength * fSex,random =~1| Farm, family = binomial, data = DeerEcervi)summary(DE.PQL)DE.PQL$coefficients[1][1]# Modellvoraussagen berechnen# So könnte direkt auf die coefficients zugegriffen werden: DE.PQL$coefficients[[1]][1]g <-0.8883697+0.0378608* DeerEcervi$CLength# Rücktransformieren aus Logitp.averageFarm1 <-exp(g) / (1+exp(g))# Sortierung der Hirschgrössen für's PlottenI <-order(DeerEcervi$CLength)# Plottenplot(DeerEcervi$CLength, DeerEcervi$Ecervi.01,xlab ="Length",ylab ="Probability of presence of E. cervi L1")lines(DeerEcervi$CLength[I], p.averageFarm1[I], lwd =3)# Vertrauensintervalle (CI) mit SD von Random factor berechnen# Generell CI = mean + 1.96*SDp.Upp <-exp(g +1.96*1.462108) / (1+exp(g +1.96*1.462108))p.Low <-exp(g -1.96*1.462108) / (1+exp(g -1.96*1.462108))lines(DeerEcervi$CLength[I], p.Upp[I])lines(DeerEcervi$CLength[I], p.Low[I])# Dasselbe mit dem lme4-Packagelibrary("lme4")DE.lme4 <-glmer(Ecervi.01~ CLength * fSex + (1| Farm),family = binomial, data = DeerEcervi)summary(DE.lme4)library("glmmML")DE.glmmML <-glmmML(Ecervi.01~ CLength * fSex,cluster = Farm, family = binomial, data = DeerEcervi)summary(DE.glmmML)```